Molecular Drug Design
Novitero Ltd. is capitalizing on the recent technological advancements in the fields of chemistry, Chemoinformatics, biology and bioinformatics and create a personalized medicine approach of new drug design and production. By utilizing the information gathered by marketed drugs and drugs under development and the corresponding resistance mutations, we develop new, improved and patented drugs, based on already marketed drugs acting on clinically validated targets and adapt their new structure to functional features bypassing multiple mutations.
The focus of the company is on drugs that are associated with treatment-emergent mutations that cause drug resistance. The novel and adapted personalized drugs that bypass multiple mutations would enable life-saving treatment of resistant patient populations with emergent mutations.
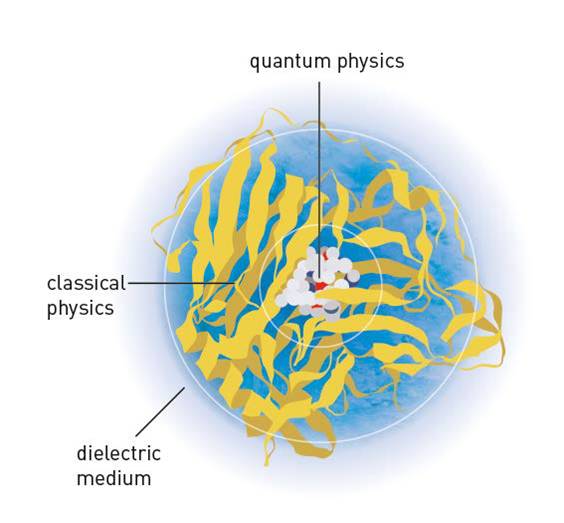
 In-silico approaches for design of new drugs apply, in part, methods and techniques in theoretical and computational chemistry which largely rely on a physical understanding of the chemical or biological system of interest, in atomic detail. These methods encompass, for example, molecular mechanical and/or quantum mechanical calculations, molecular dynamics simulations, and free energy calculations (e.g. for prediction of binding affinity). The great importance of developments in this discipline was recently recognized by the 2013 Nobel Prize in Chemistry – awarded jointly to Martin Karplus, Michael Levitt and Arieh Warshel, “for the development of multiscale models for complex chemical systems”.
In-silico approaches for design of new drugs apply, in part, methods and techniques in theoretical and computational chemistry which largely rely on a physical understanding of the chemical or biological system of interest, in atomic detail. These methods encompass, for example, molecular mechanical and/or quantum mechanical calculations, molecular dynamics simulations, and free energy calculations (e.g. for prediction of binding affinity). The great importance of developments in this discipline was recently recognized by the 2013 Nobel Prize in Chemistry – awarded jointly to Martin Karplus, Michael Levitt and Arieh Warshel, “for the development of multiscale models for complex chemical systems”.

However, while these methods constitute the core theoretical framework underpinning most modern molecular modeling tools (e.g. docking), they are often not applicable for large-scale datasets of compounds, such as those used for virtual screening procedures, due to limited computational resources. Empirical methods such as quantitative structure-activity relationships (QSAR) fill this void by identifying statistical patterns in existing observed data, and using them to predict and guide the design of future active compounds. These methods are generally less computationally-demanding, and thus are more adapted for large volumes of data. Empirically-based methods may be generated together with physically-based methods at different stages of the virtual screening process. In particular, QSAR models quantitatively correlate the physico-chemical features derived from the chemical structure of the molecules with the biological activity, and then used to optimize the active compounds in order to improve or modify the desired biological function. Therefore, they could be beneficial for lead optimization. Alongside with pharmacophore modeling, QSAR is, indeed, one of the most widely used tools in ligand-based drug design.
Different from high throughput or combinatorial chemistry approaches, often used for the development of single-mutant-selective drugs, in our studies we combine computational chemistry and molecular dynamics for the design and selection of mutated proteins and subsequently computer-aided drug design, de-novo ligand design and scaffold hopping technologies for the re-design of mutation-bypassing compounds. The technology, previously not employed for cancer therapeutics, is markedly influenced from knowledge gained the anti-HIV field.
The compounds designed by our experimental approach would pioneer the novel approach to develop drugs addressing multiple mutations and would enable boarder therapeutic coverage for cancer.
We hope that these drugs and novel approach would become an important milestone in rendering cancer a chronic, manageable or curable disease.